Mag. Dr. Rainer Hubmann
E-Mail: rainer.hubmann@meduniwien.ac.at
T: +43 (0)1 40400-52830
T: +43 (0)1 40400-44140 (assistant and lab)
Dr. Rainer Hubmann studied genetics at the Vienna Biocenter (VBC)/Max Perutz Labs of the University of Vienna, where he completed his diploma thesis in the Hirth group in cooperation with Kim Nasmyth (IMP) as a member of the Vienna Cell Cycle Club. He has been at our department since 1997, first as a PhD student (Ludwig Boltzmann Institute for Cytokine Research), postdoc (Comprehensive Cancer Center Vienna/Drug & Target Screening Unit; Ludwig Boltzmann Cluster Oncology), and currently as senior scientist/principal investigator (PI), where he also serves as vice-coordinator of the CCC research cluster "Microenvironment, Vasculature & Metastasis". He has published several papers as first author in renowned journals, patented therapeutically and diagnostically useful findings, and successfully submitted numerous research projects.
His scientific focus is on research into the development of leukemias/tumors with a focus on NOTCH2 signal transduction. The morphogenic stem cell factor NOTCH2 plays a central role in embryogenesis and adult tissue homeostasis and is deregulated as a tumor-inducing oncogene in many leukemias and tumors. NOTCH2 therefore represents a promising target for therapeutic interventions with curative potential. Using promoter analyses, Dr. Hubmann was able to show that NOTCH2 is responsible for the overexpression of the CD23 surface marker in CLL. As part of this project, he established a NOTCH2 electrophoretic mobility shift assay (EMSA) as a drug screening tool and used it to identify the Aspergillus antibiotic gliotoxin as a potent, therapeutically relevant nuclear NOTCH2 inhibitor. Gliotoxin is highly efficient in killing neoplastic cells in cell culture via the induction of programmed cell death (apoptosis) and significantly inhibits tumor growth in a melanoma mouse model.
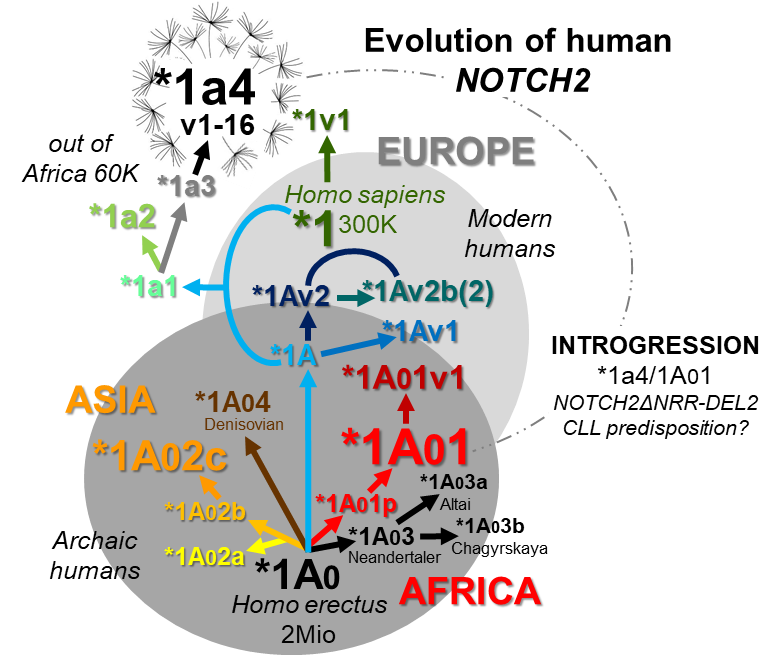
Dr. Rainer Hubmann is currently investigating the genetic cause of the deregulation of NOTCH2 signal transduction in CLL. His preliminary results show that CLL cells express a constitutively active NOTCH2 form that lacks the so-called "NOTCH2 negative regulatory region" (NRR). The reason for this is probably a recombination between paternally and maternally inherited, incompatible NOTCH2 gene variants. This physiological repair mechanism could be triggered by a DNA double-strand break in a CLL progenitor cell and thus represent the initial "malignant hit" in CLL pathogenesis. Genome analyses show that highly complex, archaic NOTCH2 "supergene" variants are centrally involved in this process (see figure). These could have been incorporated into the gene pool of modern humans in the course of evolution through mating (introgression). The data should not only make an important contribution to understanding the development of NOTCH2-associated neoplasias, including familial and geographical aspects, but also form the basis for prognostically and diagnostically relevant test procedures.